Chem406:Biophysical Chemistry
Assignment For Lab 3-Jmol
Due - Wednesday, September 27, 2005 at 5:00pm
Introduction
In this assignment you will explore the structure of a protein using Jmol. Each of you will be assigned a different protein. You will prepare a series of scripts, test them, and submit them for evaluation. Scripts are files that can be loaded into Jmol to execute a series of commands and regenerate a scene. You will also write a brief description of the protein, its interaction with the hetero group, and some of its structural features.
Each of you will work with a different protein. All the assigned proteins contain a hetero group, which comprises a cofactor, prosthetic group, or inhibitor. Obtain your structure file from the Protein Data Bank.
Your instructor will demonstrate how to make your script files with BBEdit and then load them into Jmol. All your script files should be named xxxx#.spt, where xxxx
denotes the four character PDB file ID for your protein, and # is a one-digit number (example: 1cyo2.spt).
Click here to obtain your assignment.
Script List
You will create a collection of five script files. When loaded into Jmol, each will recreate one of the scenes described below. Once you have constructed a described scene, use rotation, translation, and zooming to a produce clear, informative view in which the features requested are clearly displayed. Use the "show orientation" command to create "moveto" command, and then include the "moveto" command in your script.
To test a script, type the following command at the prompt in the Rasmol Script console window:
Command line: script xxxx#.spt < return >
For this to work properly, the Jmol.jar file, your pdb file and the script file should all be in the same folder.
After a delay of a few seconds, you should see the script you created.
If your protein contains two or more identical
chains, restrict all views to one chain and its associated hetero
group. If there are hetero groups in addition to the one listed in
the table of assigned files, do not include them with any of the
requested views. Include only the hetero group listed in the table.
Finally, do not leave water molecules on display in any of your
completed views.
Here are the script files you should make:
xxxx1.spt
Backbone display of the protein, colored by structure; spacefill display of hetero group(s), colored CPK.
xxxx2.spt
Closeup of the hetero group(s) alone, in ball & stick display, with shadows and specular reflections, colored CPK.
xxxx3.spt
Closeup of ligand binding, showing the ligand along with all protein atoms that are are part of an amino acid sidechain that contains at least one hydrogen bond or salt bridge to the ligand as sticks display (wireframe 0.3), colored CPK; and wireframe display of the surrounding protein, colored CPK. If any of the hetero atoms disappear in sticks display (this often happens with metal ions), select them individually and display them in ball & stick. In addition, use distance monitors to display hydrogen bonds or salt bridges between hetero atoms and
protein atoms. Find as many such interactions as you can. Arrange the view so that the interactions and distance labels are in clear view.
CRITERIA FOR IDENTIFYING HYDROGEN BONDS AND SALT LINKS: Remember that hydrogen bonds involve two electronegative atoms that share one hydrogen atom. In proteins, hydrogen bonds involve mostly N and O, but never C. Hydrogen atoms are not visible in electron density maps from protein crystallography, so they are not shown in protein models. Therefore, we must infer the presence of a hydrogen bond when we find a pair of appropriate atoms within about 2.8 angstroms of each other. In addition to chemical criteria (one atom must be of a type that is normally protonated, one atom must be N or O, and the other must be N, or O) and distance criteria (2.8 ± 0.5 angstroms), the atoms must satisfy geometric criteria for hydrogen bonding. The hydrogen is involved with a bonding orbital on one atom, and with a nonbonding orbital on the other, so a line extending from one hydrogen-bonded atom to another must make either a tetrahedral or trigonal planar angle with the other covalent bonds at both atoms. Salt bridges, or electrostatic interactions, must involve atoms of opposite charge. The atoms can be in almost any orientation to each other, at distances near the van der Waals contact distance of about 3 Å.
Tips: To identify atoms on the protein that are potentially hydrogen bonded to the ligand, select nitrogen and oxygen atoms that are within 3-Angstroms of a nitrogen or oxygen on the ligand. This can be done using "within" functions to do selections, For example
select (*.N?? or *.O??) and protein and within(3.0,((*.N?? or *.O??) and ligand))
In plain English this reads "select any nitrogen or oxygen atom that is also in the protein and that is also within 3.0 Angstrom of any nitrogen or oxygen that is also in the ligand".
After you have selected these atoms render them, for example, as spheres with radii that are 20% of the vander Waals radiius (spacefill -20). Double-click on each atom and use the picking monitor to find the nitrogens and oxygens on the ligand that are within 3.0 Angstroms. By then double-clicking on the atoms you find within 3.0 Angstroms, you can leave the monitor in place. This is what it should look like.
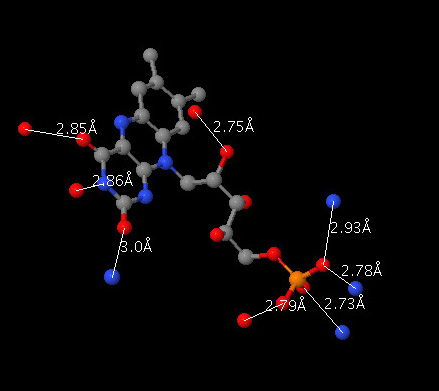
To include the distance monitors in your scripts, use the command "monitor atomno1 atomno2", where atomno1 and atomno2 are the atom numbers for the two atoms that you wish to connect. You can find the atom number for an atom by clicking on it and looking in the console; the atom number is the integer the follows the # sign.
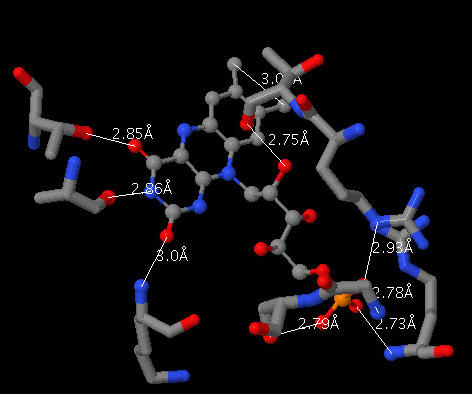
To select all of the atoms in a sidechain that contains at least one atom from the previous select, nest it within a second within functions:
select within(group,(*.N?? or *.O??) and protein and within(3.0,((*.N?? or *.O??) and ligand)))
In plain English this reads "select any atom in within the same amino acid (group) as any nitrogen or oxygen atom that is also in the protein and that is also within 3.0 Angstrom of any nitrogen or oxygen that is also in the ligand".
If we use "wireframe 0.3" to render this selection it should look something like this:
xxxx4.spt
Display all the atoms in the ligand along with all of the atoms on the protein that are within 6.5 Angstroms of the ligand. Render this selection as spheres with vander Waals radii. Color the carbon atoms (only) of ligand green, and all other atoms, including non-carbon atoms of the ligand, in CPK colors. Arrange this picture conveniently for slabbing through the structure to show the fit of the hetero group into the protein. If the slabbing is not working, try turning it on with the command "slab on".
xxxx5.spt
Ball & stick display of one prominent element of secondary
structure: either a pleated sheet of least three strands, or one
alpha helix of at least 8 residues. Main chain atoms colored CPK.
Side chains colored as follows: polar green, positive blue, negative
red, hydrophobic yellow. Mainchain H-bonds displayed. Note whether
the sheet or helix in your display appears to be amphipathic -- that is, having one side hydrophilic and one side hydrophobic. Then note whether the sheet or helix is buried within the protein, or on its surface. In your written description of the protein (see below), comment on the distribution of nonpolar and polar groups and tell how they fit the location (surface or buried) of the sheet or helix in the protein.
Description of the Protein
To complete this assignment, write a brief description (no more than one page, double-spaced) of your assigned protein, referring to the scenes that are recreated by you scripts. In the first paragraph, give the name and source of the protein, and then describe its function. If it is an enzyme, give the reaction that it catalyzes, drawing molecular structures of substrate(s) and product(s).
In the second paragraph, describe the structure, including
- the number of chains and number of residues in each; and
- major secondary structural elements, and the residue numbers that they span.
In this paragraph, be sure to include the description described above for xxxx5.spt.
In the third paragraph, describe the binding of the hetero group to the protein, referring to the appropriate scripts.
Model your written description after the introductory description of any protein (for example myoglobin) in a biochemistry textbook. Write this description with a Microsoft Word, print it, and also save it in the same folder as your scripts. You should name the description file username_lab3.doc. You should make a new folder that contains copies of the following items:
- username_lab3.doc
- xxxx1.spt
- xxxx2.spt
- xxxx3.spt
- xxxx4.spt
- xxxx5.spt
- xxxx.pdb
Name the folder username_lab3 and create an archive file (username_lab3.zip) from the folder
When you have completed the assignment, email me with your username_lab3.zip as an attachment The assignment is due by 5:00pm on Wednesday, September 27..
Here are some sources of information about your protein and its hetero group:
- Look up the protein and its hetero group in the index of a Biochemistry textbook.
- Click web links in the header of the PDB file to see PubMed abstracts about your protein.
- Using PubMed, search for recent papers about your protein.
- Explore other links accessible from the Summary page for your protein at the Protein Data Bank.
Protein Assignments:
Name
|
PDB ID
|
Hetero Group (Ligand)
|
| Shanna Berger |
|
Flavin mononucleotide (FMN)
|
| Stephanie Booms |
|
AP5A (ATP-analog
inhibitor)
|
| Mike Brunetto |
|
S-adenosylmethionine
|
| Amy Groswell |
|
Coenzyme A
|
| Mike Davey |
|
Thiamine Pyrophosphate
|
| Bret Deml |
|
Pyridoxal Phosphate
|
| Michael Fredericks |
|
Vitamin B12
|
| Russell Garton |
|
Folic Acid
|
| Chris Groen |
|
Flavin mononucleotide (FMN)
|
| Andy Lee |
|
Biotin
|
| Fong Lee |
1ai2
|
Nicatinamide adenine dinucleotide phosphate (NADP) |
| Cassie Pherson |
1trk
|
Thiamin Pyrophosphate |
| Venay Rao |
1zin
|
AP5A (ATP-analog inhibitor) |
| Mitch Springer |
|
Thiamine Pyrophosphate
|
| James Thomas |
|
|
| Jim Zook |
|
Pyridoxal Phosphate |
| Jay Zuehlke |
|
Flavin mononucleotide (FMN) |
updated: Monday, October 9, 2006