The thermodynamics of each enzyme plays an important role for stabilization and describing if the reaction is favorable. The entropic and enthalpic energies help to stabilize the transition state formed by chalcone isomerase by allowing three water molecules to be expulsed pushing the entropic advantage towards the chalcone isomerase. Also the release of the enzyme strain reduces the activation energy (4). The free energy found for mandelate racemase is negative showing a favorable reaction. Not everything in the reaction is favorable, however. The enthalpy for conversion of the substrate to product is positive showing the process is unfavorable enthalpically. Also the entropy change with the substrate binding is negative making the process unfavorable entropically (12). The hydrogen bonding with Glu317 and Asn197 are polar interactions, which is a major contributor to the enthalpy of the transition state stabilization. Also the electrostatic interactions between the hydrophobic pocket and the phenyl group contribute free energy to the ground state binding. Other sources for free energy that come from the binding substrate in the hydrophobic cavity include: entropically driven hydrophobic interactions, specific electrostatic interactions, Van der Waals forces and dehydration of the pocket. Another source of free energy is the release of ordered water molecules. All of these interactions included in binding and the reaction help to provide free energy and favorable processes, however, most isomerases have free energies close to zero because their reactants and products are very similar. No thermodynamic information could be found about DNA topoisomerase 1. The interactions and source of energy is important to catalyze the enzyme’s reaction.
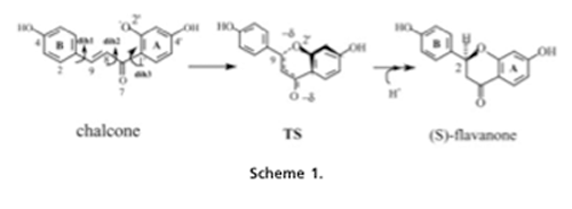
All three enzymes have similar types of reactions, but involve different substrates, mechanisms and active sites. Chalcone isomerase has two substrates, chalcone and flavanone. The active site molds the substrate for reactive conformation. The contact between the O2 of the substrate and the C9 of the active site is a prerequisite for the chalcone,flavanone reaction. There are two schemes proposed for the mechanism of the reaction. Scheme two includes an intermediate (Figure 2), where scheme one considers the intermediate as a transition state (Figure 1.a). The transition state is stabilized by the hydrogen-bonding network formed by the Thr43, Tyr106 and ketone O7. There is also a partial formation of the C9-O2’ bond allowing the transition state to fit into the cleft formed by the active site. This formation is an interaction accompanied by a proton transfer from a protonated amino group of Lys95 to the transition state O7. Also during this state three tightly bound water molecules are released and the active site becomes open. The narrow shape of this state exposes the hydrophobic residues. The different interactions involved in moving chalcone to the transition state is what drives the reaction. The transition state also increases the rate of catalysis. The reaction has many aspects that cause it to move forward and work. Electrostatic stabilization comes from the chalcone O7 approaching the Lys97 protonated amino group. Also the movement and change of shape of the ligand induces the expulsion of the three water molecules. The backbone structure also changes, so the hairpin loop recedes away from the ligand-binding pocket during the formation of the transition state (9). The last important action of the reaction catalysis is the polarization of the bound water molecule by the side chain Tyr106 followed by the water-mediated deprotonization of the 2’-hydroxyl group of the chalcone, formation of the oxyanion and the intramolecular attack on the alpha,beta unsaturated double bond (4) (Figure 1.b).
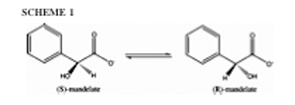
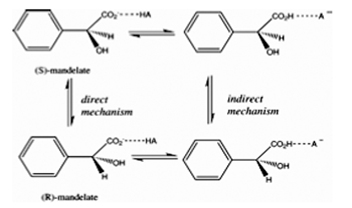
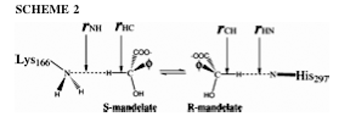
Mandelate racemase also has two schemes for the mechanism of the reaction. Scheme one involves an intermediate (Figure 2.a) and scheme two refers to the intermediate as the transition state (Figure 3). The substrates for both schemes are (S) and (R)-mandelate. These two are para-substituted mandelic acids. For both schemes Lys164, Glu317 and Mg2+ are involved in the ligand binding. Also the electron-withdrawing phenyl substituents enhance the enzyme activity. An aromatic system must be present in the beta position for the ligand to bind (3). Also the aromatic ring of the ligand must be within a large hydrophobic cavity at the mouth of the active site. This hydrophobic cavity solvates the phenyl group of the substrate (11). The second scheme also must have an interaction between Lys166 and Asn197 to change the conformation of the enzyme. The intermediate found in scheme one is an enolic intermediate. Electron density withdrawal stabilizes the intermediate when proton abstraction takes place (3). Another stabilizer is the enzyme-ligand interactions as well as enhanced hydrophobic interactions. The phenyl group also creates an electron sink to stabilize the intermediate. The steric constraints of this intermediate form allow the enzyme to be more stringent than the substrate complex (11). In scheme two the transition state is seen as the substitute for the intermediate. In this state the ligand seems to be more product like and there seems to be a small energy barrier. Also the active site has a maximum of negative charge concentrated on the substrate that the enzyme has to withdraw. The Mg2+ ion involved in the reaction is shown to attract that negative charge during the whole reaction and to stabilize the transition state (3). In the intermediate state of the first scheme the Asn197 interacts with the alpha-OH of mandelate to facilitate stabilization of the altered substrate. The two different schemes work differently because scheme one is a three-step mechanism (Figure 2.b) and scheme two is a direct mechanism. Both catalyze the rapid carbon-hydrogen bond cleavage and use Mg2+ assisted abstraction of the alpha-protons of carboxylic acid. Also stabilization of a negative charge is needed for the reaction. In scheme one an amino group of Lys166 is a general base catalyst that abstracts the alpha-proton from (S)-mandelate. The imidazole group of the His297 is used as the general base catalyst that removes the corresponding alpha-proton from the (R)-mandelate. This scheme also uses the alpha-OH group for binding of the substrate (3). Another important amino acid is Glu317, which is used as a general acid catalyst (11). There are three basic steps of scheme one. The first step is the catalytic protonation of the carboxylate group in the mandelate substrate by the hydrogen-bonded residues Lys164/Glu317 (Figure 4). The next step is the configuration inversion that takes place through the deprotonation of mandelic acid by Lys166 and the back-donation by His297. The last step in the mechanism is the back-transfer of the catalytic proton of the carboxylic group to the Lys164/Glu317 (3). Scheme two has a different approach where there is no intermediate. This means there is no protonation of the mandelate carboxylate prior to proton abstraction. This scheme also makes the reaction more symmetric. The mechanism is simple, where the Lys166 pulls up on the alpha proton from (S)-mandelate substrate while protonation by His297 converts the intermediate into (R)-mandelate. This is a one step process. In order for this process to take place the Lys166 must play a major role in the first part of the acid/base catalysis, while His297 plays the major role in the second part of the acid/base catalysis. It is also proposed that the Asp270 forms a catalytic dyad with His 297 to modulate the pK (11).
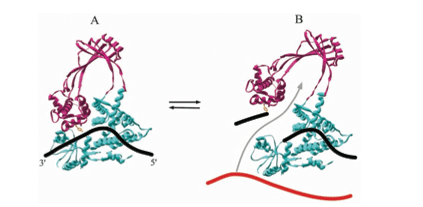
DNA topoisomerase 1 has one substrate, which is the DNA double helix. The enzyme binds to a single strand of DNA (13). There are covalent attachments to the 5’ end of the broken strand, and the enzyme is tightly clamped to the DNA by disulfide bonds. This reaction removes supercoils in the DNA one at a time. This is done by removing the torsional stress (6). The reaction also changes the topology of the DNA. The enzyme is bound by the nicked DNA to prevent the DNA from swiveling and also to allow passage of the second DNA strand through the enzyme-bridged gate before resealing. This allows for more controlled topological changes. The relaxation that occurs through DNA topoisomerase 1 is the relaxation of negative supercoils. The DNA is converted to a twisting configuration to promote enzyme binding. The topoisomerase also has to move the DNA against a force to complete the reaction (6). Also a rotation of the DNA helix must occur downstream from the cleavage site before the reaction can begin. The mechanism for this catalyzed reaction is that one strand of DNA is broken to allow rotation of one region of the duplex. A nucleophilic attack occurs by the tyrosine in the active site on the DNA phosphodiester bond. This causes the cleavage in the strand. Then the formation of the phosphodiester bond between the tyrosine and the 3’-end of the strand enables the resealing of the DNA by the reversal of the cleavage reaction. There are no known intermediates for this reaction. Also no cofactors or cations are involved in the reaction. After the reaction the enzyme must undergo a conformation change to accommodate for the rotation of the DNA (6) (Figure 6).