Amino acids are the building blocks for proteins, but they are also precursors for a number of other important biomolecules (e.g. melanin, thyroxine, adrenaline). Excess amino acids in the diet (derived form protein digestion) are not stored but are broken down. The nitrogenous part is converted into urea for excretion while the carbon skeletons may be used in various ways. In principle, the carbon skeletons are ultimately oxidized to provide energy. However, it is more unusual to speak of amino acids being ketogenic or glucogenic on the basis of whether their carbon skeletons are broken down via acetate (acetyl-CoA), and would therefore be capable of producing ketosis, or whether they might be converted into glucose via gluconeogenesis. Some amino acids are both ketogenic and glucogenic.
Humans can synthesize about half of the 20 amino acids needed for protein synthesis: the rest need to be supplied in the diet. Thus it is said that some amino acids are essential and some are nonessential. When proteins are being synthesized, an adequate supply of all 20 amino acids must be present. The system cannot 'wait' until an amino acid turns up and nor can it continue in the absence of one of the amino acids.
The body therefore needs to operate its amino acid economy very carefully. In the absence of a storage mechanism, it must bring together all 20 amino acids in the cell cytosol for protein biosynthesis, synthesizing those it can, if necessary, and obtaining the others in the diet. It must also have pathways for disposing of the amino acids. For an adult in nitrogen balance an amount of amino acids equal to the entire dietary intake is degraded each day. Thus, in the metabolic map there are a great number of pathways for dealing with amino acids (biosynthetic, degradative, transaminating, deaminating) and a correspondingly large number of enzymes. This gives a great potential for genetic diseases. A mutation affecting just one enzyme in just one amino acid metabolizing pathway can have very severe or even fatal results.
There are indeed very many inherited diseases involving enzymes of amino acid metabolism (see Appendix 4). In the past they have been diagnosed by biochemical criteria, but in the future we may expect individuals at risk to be identified by prenatal diagnosis. Eventually we may see gene and enzyme-replacement therapy for treating diseased individuals in place of the dietary restriction treatments which are all that can be achieved at present.
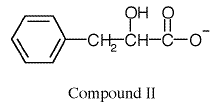
A male child, Simon V., had been born after a normal pregnancy but thereafter had failed to develop normally. When seen at age 10, he was unable to feed himself, could say only a few words and displayed involuntary limb movements. His IQ was 65. He had light blue eyes, light-colored skin and his hair was white in places. His urine had a musty odor. Large amounts of an amino acid were present in the urine. In addition a compound (compound I) was isolated from the urine in substantial amounts which when incubated with NADH and lactate dehydrogenase (LDH), gave rise to following compound (compound II):
* This Case study is taken from Higgins, S.J., Turner, A.J. and Wood, E.J., Biochemistry for the Medical Sciences., Longman Scientific and Technical, 1994.
![[UWEC Web]](../../Media/smuwec.gif) [Colleges & Dept's] [Chemistry Dept.]
[Colleges & Dept's] [Chemistry Dept.]
Warren Gallagher
Department of Chemistry
(715) 836-5388
wgallagh@uwec.edu
updated: